AIMD-AMIDURES
Nom du projet
Modélisations de type ab initio molecular dynamics (AIMD) des réactions de défluoration et d’amination de fluoropolymères (téflon et nafion) par des alkylamidures
Acronyme du projet
AIMD-AMIDURES
Nom et prénom du porteur du projet
Guillaume Herlem
Contacts
guillaume.herlem at univ-fcomte.fr
fabien.picaud at univ-fcomte.fr
Etablissement
Université de Franche-Comté
Laboratoire
Nanomédecine (jusqu’au 31.12.2023), puis SINERGIES (à partir du 01.01.2024)
Equipe de recherche
Ingénierie de la Santé
Résumé du projet
Le projet AIMD-AMIDURES vise à étudier théoriquement, sur la base de calculs de type AIMD (ab initio molecular dynamics), le mécanisme réactionnel entre un sel d’alkylamidure de lithium ou de sodium (ou azanure de lithium ou de sodium officiellement dans la nomenclature IUPAC) et un fluoropolymère. En effet, les fluoropolymères peuvent ainsi être défluorés et aminés selon une découverte récente et brevetée https://data.inpi.fr/brevets/FR3130277?q=herlem#FR3130277. Le modèle retenu pour mener à bien ce projet est la réaction entre l’amidure de 2-aminoéthyl de sodium (NH2-CH2-CH2-NHNa) et le polytétrafluoroéthylène ou PTFE (chaîne infinie de -CF2-), solvatés par l’éthylènediamine, dans une boîte périodique.
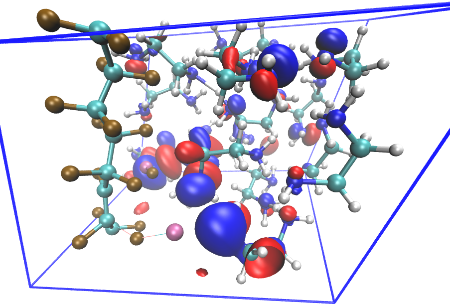
Objectif initial du projet
Simulations et traitements réalisés
Le code dans ce travail est CP2K https://www.cp2k.org/. En effet, il est parallélisable, mis à jour régulièrement et différents outils peuvent compléter ces calculs pour comparer des mesures spectroscopiques aux simulations théoriques (code TRAVIS, tool for analyzing and visualizing trajectories from all kinds of Molecular Dynamics or Monte Carlo simulations.http://www.travis-analyzer.de). Le code PACKMOL https://m3g.github.io/packmol/index.html a été utilisé afin de créer un point de départ pour les simulations de dynamique moléculaire en regroupant les molécules solvatées par l’éthylènediamine. Le regroupement garantit que les interactions répulsives à courte portée ne perturbent pas les simulations. Le code Volume guesser https://m3g.github.io/packmol/nmols.shtml calcule le volume du système contenant un nombre donné de molécules de différents types afin que la densité finale ait une valeur souhaitée.
Résultats obtenus
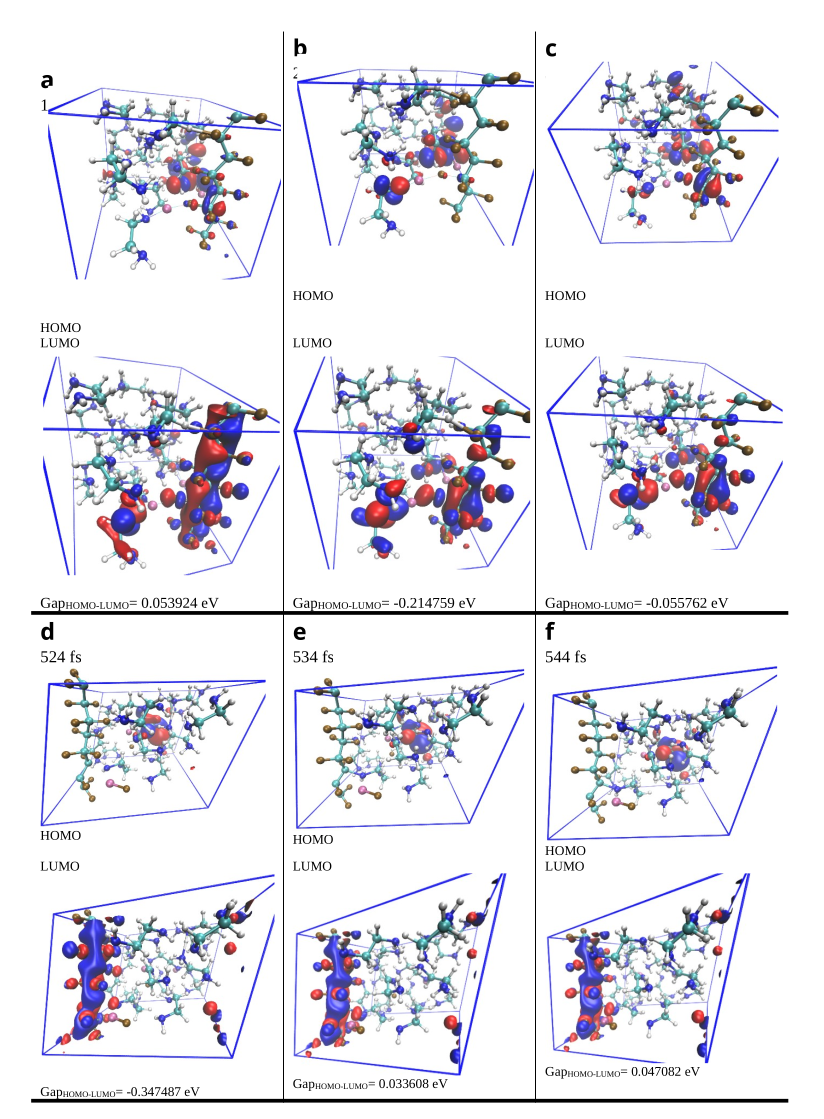
Valorisation des résultats
Comment avez-vous publié et communiqué vos résultats ?
Sous forme de brevets pour l’instant (une extension du brevet cité à l’Europe et aux USA est en cours). Des études structurales expérimentales (DRX, RMN) sont nécessaires pour aboutir à un article à haut facteur d’impact.
Valorisation dans le cadre pédagogique ?
Pas pour l’instant sauf en formation doctorale auprès des étudiants en thèse sous ma direction.
Les données ont-elles été ouvertes ?
Non car sous le sceau du brevet et de la création d’entreprise. Les débouchés de ce type de réaction sont importants dans les secteurs du stockage d’énergie, de la synthèse asymétrique et des biotechnologies. J’ai été incubé par l’incubateur DECA-BFC en Novembre 2024 à ce titre.